The Neighborhood of the Spike Gene Is a Hotspot for Modular Intertypic Homologous and
Nonhomologous Recombination in Coronavirus Genomes
Coronaviruses (CoVs) have very large RNA viral genomes with a distinct genomic architecture
of core and accessory open reading frames (ORFs). It is of utmost importance to understand
their patterns and limits of homologous and nonhomologous recombination, because such events
may affect the emergence of novel CoV strains, alter their host range, infection rate,
tissue tropism pathogenicity, and their ability to escape vaccination programs. Intratypic
recombination among closely related CoVs of the same subgenus has often been reported;
however, the patterns and limits of genomic exchange between more distantly related CoV
lineages (intertypic recombination) need further investigation. Here, we report
computational/evolutionary analyses that clearly demonstrate a substantial ability for CoVs
of different subgenera to recombine. Furthermore, we show that CoVs can obtain—through
nonhomologous recombination—accessory ORFs from core ORFs, exchange accessory ORFs with
different CoV genera, with other viruses (i.e., toroviruses, influenza C/D, reoviruses,
rotaviruses, astroviruses) and even with hosts. Intriguingly, most of these radical events
result from double crossovers surrounding the Spike ORF, thus highlighting both the
instability and mobile nature of this genomic region. Although many such events have often
occurred during the evolution of various CoVs, the genomic architecture of the relatively
young SARS-CoV/SARS-CoV-2 lineage so far appears to be stable.
Major findings:
Core ORFs undergo homologous recombination at the species, subgenus and genus levels.
CoVs can obtain AOFs through non
homologous recombination, even from other viruses or
hosts.
Recombination events are mostly localized at the Spike neighborhood.
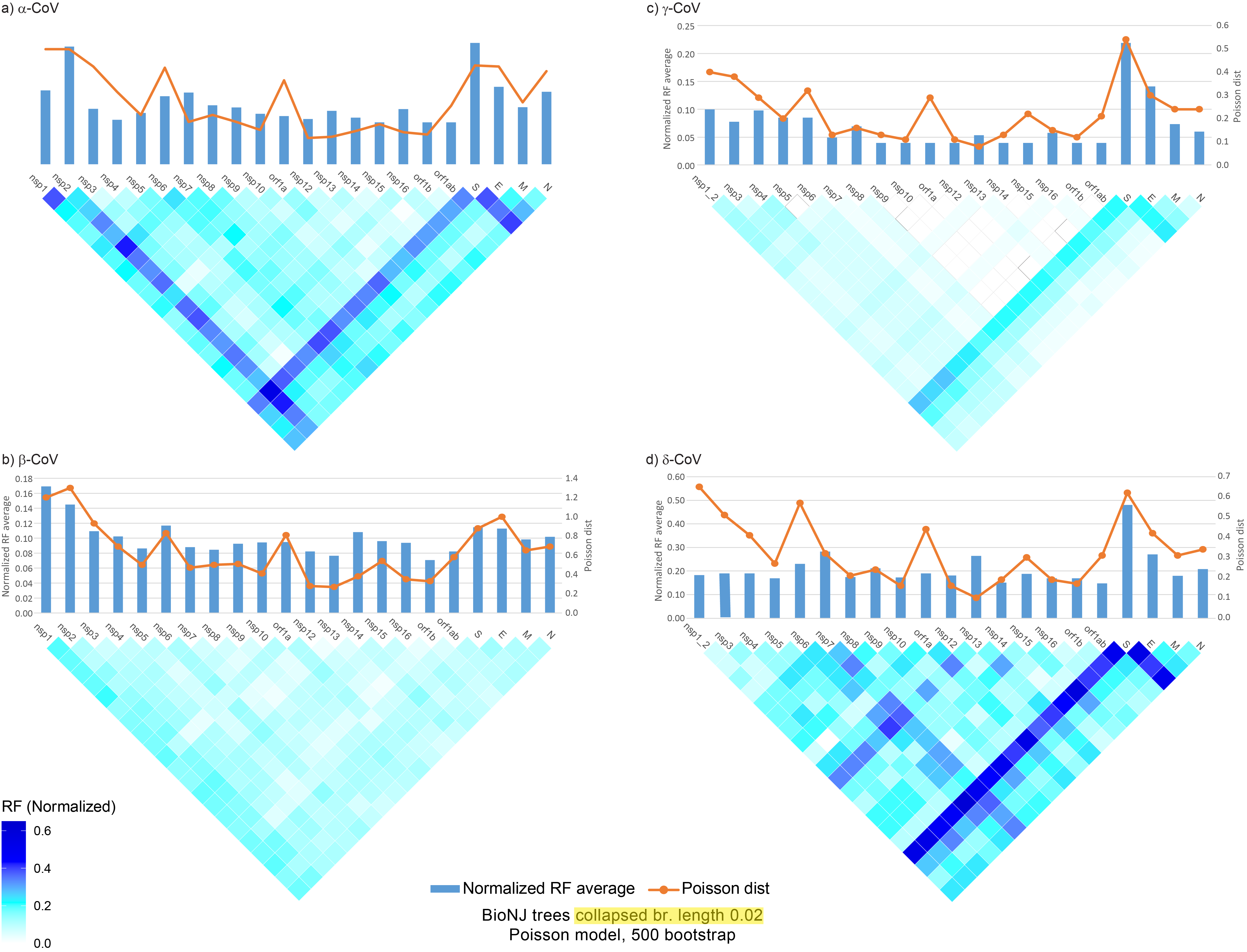
Figure 1. Matrices of incongruence among the core genomic regions of the four CoV
genera (A–D) based on the normalized RF method, for unrooted trees (calculated with
the TreeCMP server). BioNJ phylogenetic trees were generated with the Poisson model
of evolution and 500 bootstrap replicates. In addition, branch lengths <0.02
were collapsed. The orange line above each matrix displays the average Poisson
distance among sequences of the same genomic region (calculated with the MegaX
software). Blue bars above each matrix display the average RF value for that
particular region (against all other regions).
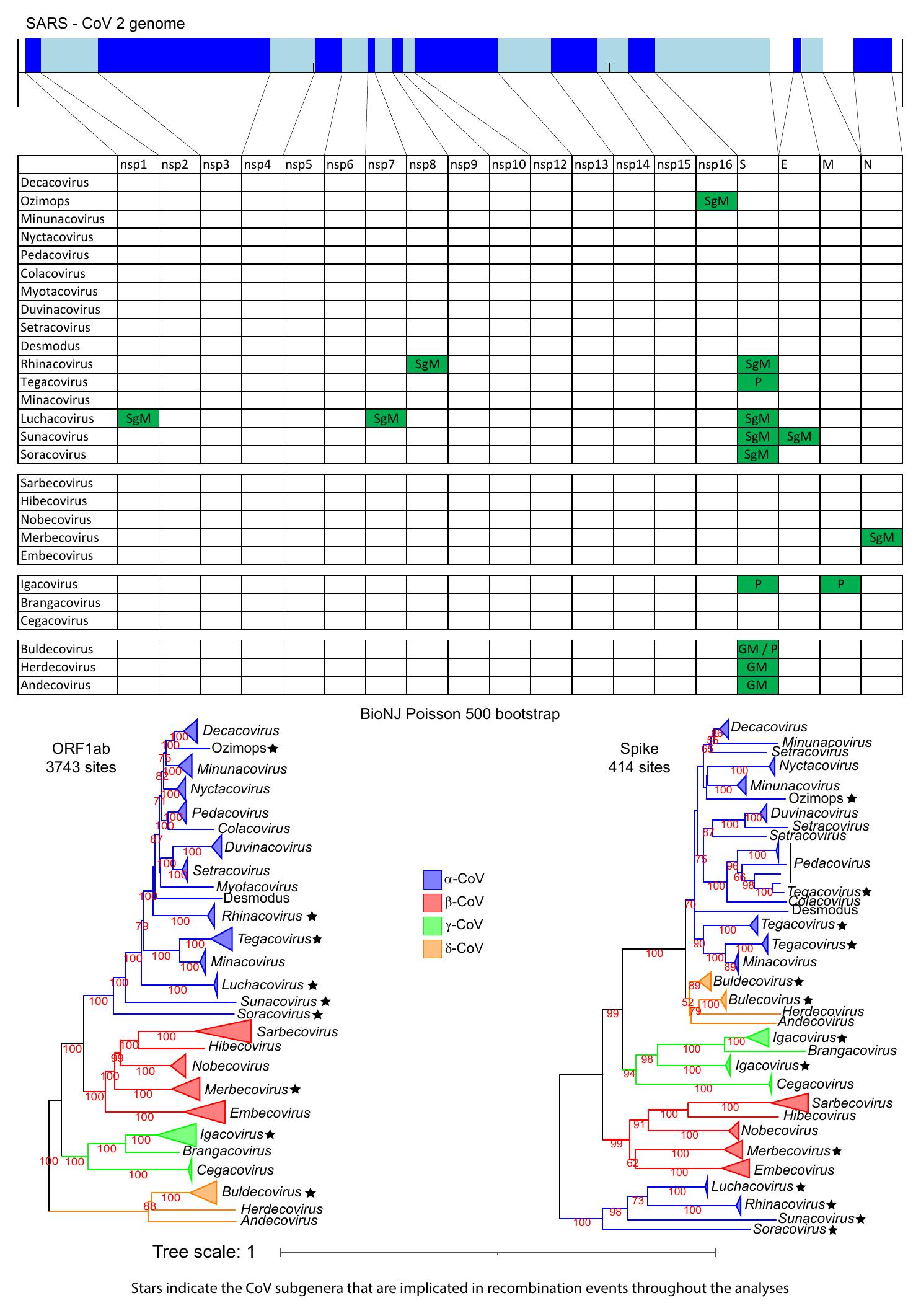
Figure 2. The genomic organization of the core ORFs and peptides of the SARS-CoV-2
genome are displayed on the top of the figure. The table/matrix below it shows which
genomic regions of the various subgenera are involved in intertypic recombination
events. “GM” represents events that occurred at the common ancestor of the genus.
“SgM” represents events that occurred at the common ancestor of the subgenus. “P”
represents more recent events that occurred for one or few members of the subgenus
and have resulted in a polyphyletic tree pattern (for that region and subgenus). All
incongruence events in the matrix are supported by the three phylogenetic tree
methods (NJ, PhyML, and Bayesian) and are also statistically significant, based on
the AU test of CONSEL. Two phylogenetic trees (of ORF1ab and Spike) for all four
genera are also included below the matrix, to visualize the recombination events of
the Spike region. In these trees, we use stars to denote subgenera that have been
involved in intertypic homologous recombination events, in any genomic region (not
only the Spike).
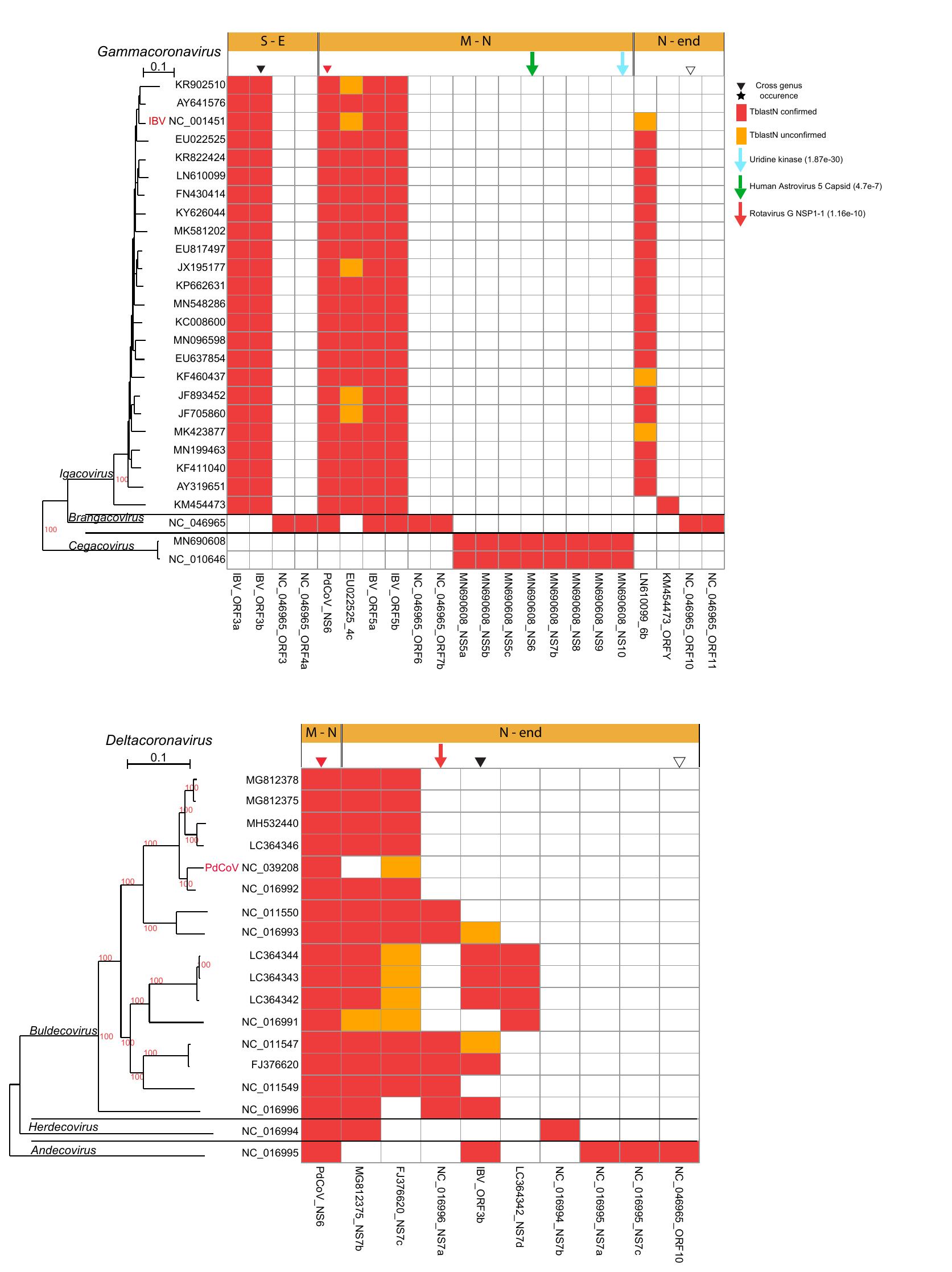
Figure 3. Presence and distribution of AOFs in the α- and β-CoVs. Each column in the
matrix represents a certain AOF. Red color (within the matrix cells) denotes the
(TblastN) presence of an AOF that is also verified by a predicted ORF with length
≥30 aa, whereas if the length of the predicted ORF is <30 aa, then it is denoted
with orange color. Stars denote AOFs that are present in both α- and β-CoV
members, whereas diamonds denote an AOF that resulted from duplication of a core
ORF. Downward arrows denote AOFs that have homologs in non-CoV genomes, together
with their best PSI-BLAST hit e-value. Horizontal orange bars (above the
matrices) denote the genomic region where the AOF is located, that is, S-E
denotes the region between the Spike and Envelope ORFs.
Figure 4. Presence and distribution of AOFs in the γ- and δ-CoVs. Each column in the
matrix represents a certain AOF. Red color (within the matrix cells) denotes the
(TblastN) presence of AOFs that is also verified by a predicted ORF with length ≥30
aa, whereas if the length of the predicted ORF is <30 aa, then it is denoted with
orange color. Inverted triangles denote AOFs that are present in both γ- and
δ-CoV members. Downward arrows denote AOFs that have homologs in non-CoV
genomes, together with their best PSI-BLAST hit e-value. Horizontal orange bars
(above the matrices) denote the genomic region where the AOF is located, that
is, M-N denotes the region between the Membrane and Nucleocapsid ORFs.
Comparative Analysis of SARS-CoV-2 Variants of Concern, Including Omicron, Highlights Their
Common and Distinctive Amino Acid Substitution Patterns, Especially at the Spike ORF
In order to gain a deeper understanding of the recently emerged and highly divergent Omicron
variant of concern (VoC), a study of amino acid substitution (AAS) patterns was performed
and compared with those of the other four successful variants of concern (Alpha, Beta,
Gamma, Delta) and one closely related variant of interest (VoI—Lambda). The Spike ORF
consistently emerges as an AAS hotspot in all six lineages, but in Omicron this enrichment
is significantly higher. The progenitors of each of these VoC/VoI lineages underwent
positive selection in the Spike ORF. However, once they were established, their Spike ORFs
have been undergoing purifying selection, despite the application of global vaccination
schemes from 2021 onwards. Our analyses reject the hypothesis that the heavily mutated
receptor binding domain (RBD) of the Omicron Spike was introduced via recombination from
another closely related Sarbecovirus. Thus, successive point mutations appear as the most
parsimonious scenario. Intriguingly, in each of the six lineages, we observed a significant
number of AAS wherein the new residue is not present at any homologous site among the other
known Sarbecoviruses. Such AAS should be further investigated as potential adaptations to
the human host. By studying the phylogenetic distribution of AAS shared between the six
lineages, we observed that the Omicron (BA.1) lineage had the highest number (8/10) of
recurrent mutations.
Major findings:
The Spike ORF consistently emerges as an AAS hotspot in all six lineages, but in Omicron
this enrichment is significantly higher.
The VoC/VoI lineage ancestors undergo positive selection, followed by purifying
selection after variant emergence.
Vaccination does not accelerate the accumulation of non-synonymous mutations at Spike.
Omicron recurrent mutations may be a result of inter-lineage recombination
(Recombination with other Sarbecovirus is rejected via CONSEL).
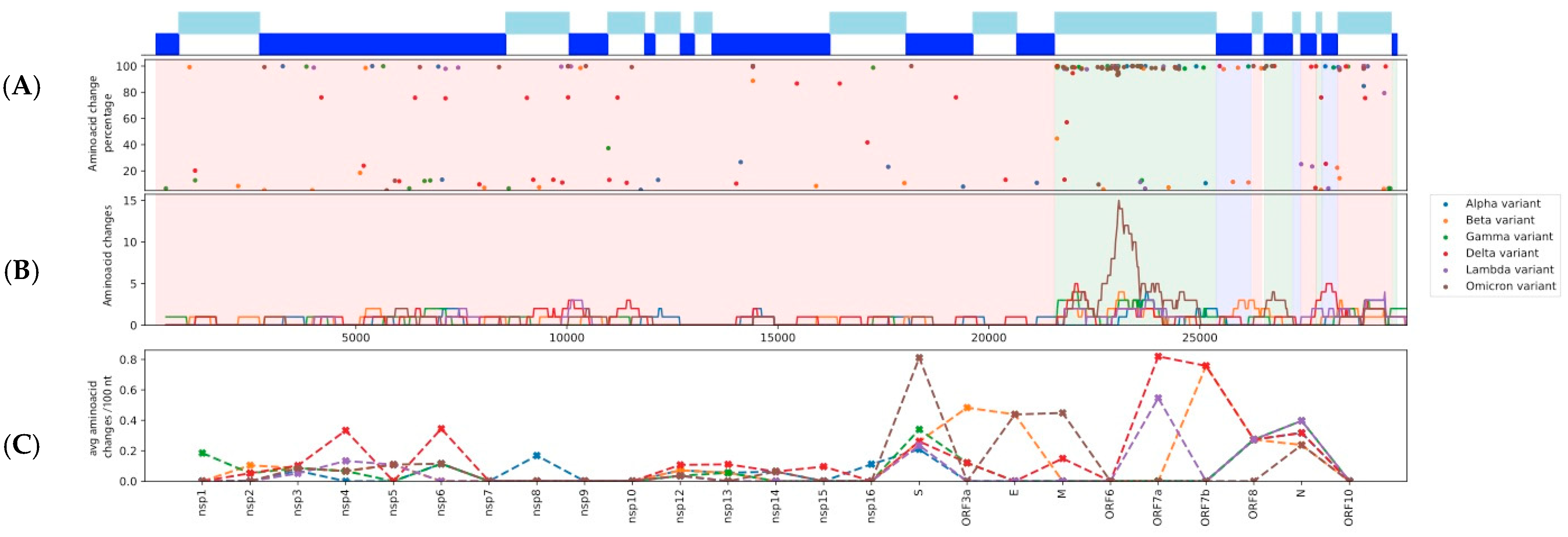
Figure 1. (A) The distribution of amino acid substitutions (AAS) across the
SARS-CoV-2 genome and their frequencies for each analyzed variant lineage.
(B) A sliding window analysis of the number of AAS for a particular region.
The size of the sliding window is 500 nt with a step of 20 nt. (C) Number of
AAS per 100 nt, for each nsp and ORF.
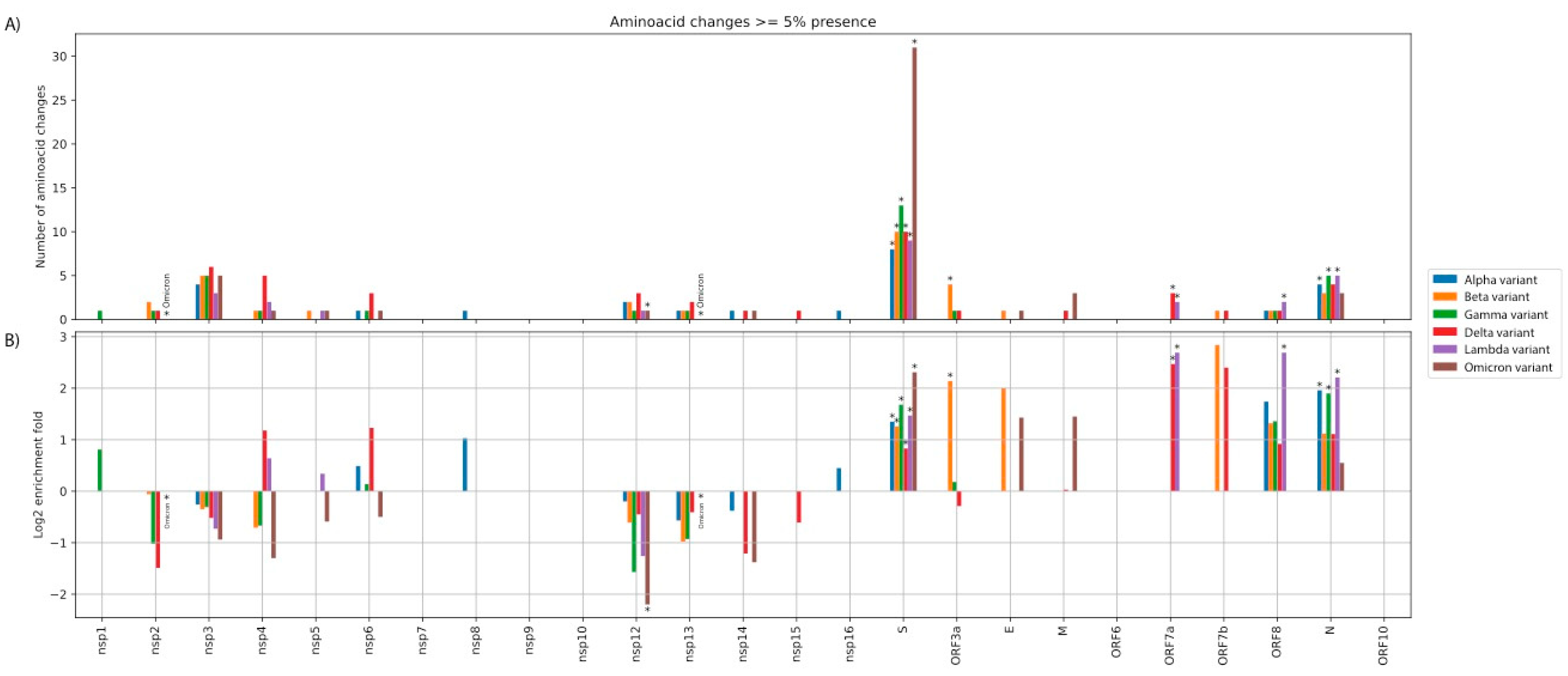
Figure 2. (A) Absolute number of amino acid substitutions (AAS) for each
nsp/ORF. (B) Log2 fold enrichment of AAS for each nsp/ORF, after
taking into
account the length of each region. Stars denote statistically significant
over/under-representation. Note that, due to the small number of AAS, several
over/under-representations may not achieve statistical significance (at p < 0.05).
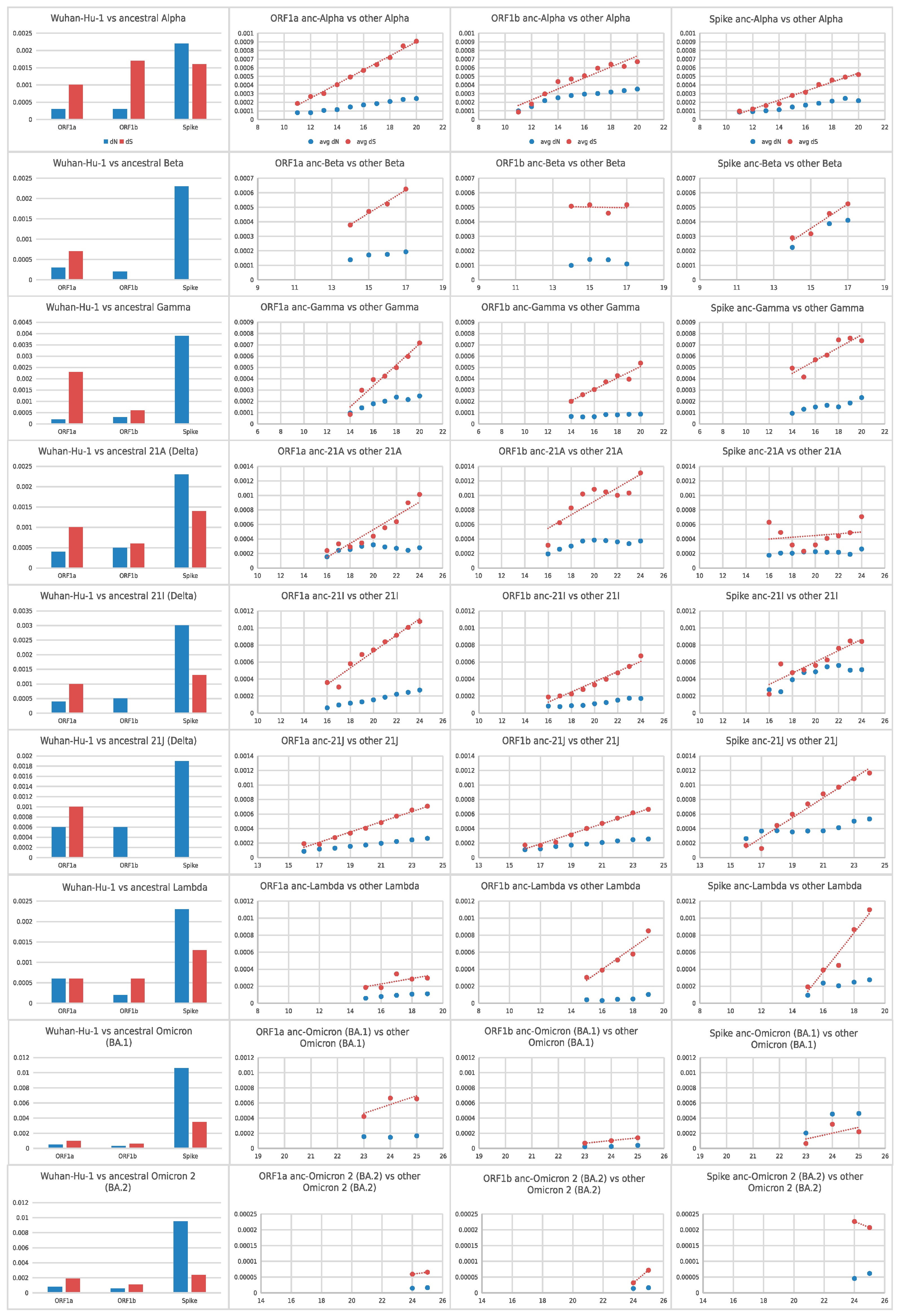
Figure 3. Cumulative average pairwise dN and dS values (y-axis values) of the
selected variant lineages, from the beginning of the pandemic (Wuhan-Hu-1) until the
ancestor of each lineage (leftmost bar-chart) and from the ancestor of each lineage
until every selected month, for ORF1a, ORF1b and Spike. The x-axis of the three
rightmost graphs for each lineage denotes the month from the beginning of the
pandemic (December 2019). Red dots denote pairwise dS values whereas blue dots
denote pairwise dN values.
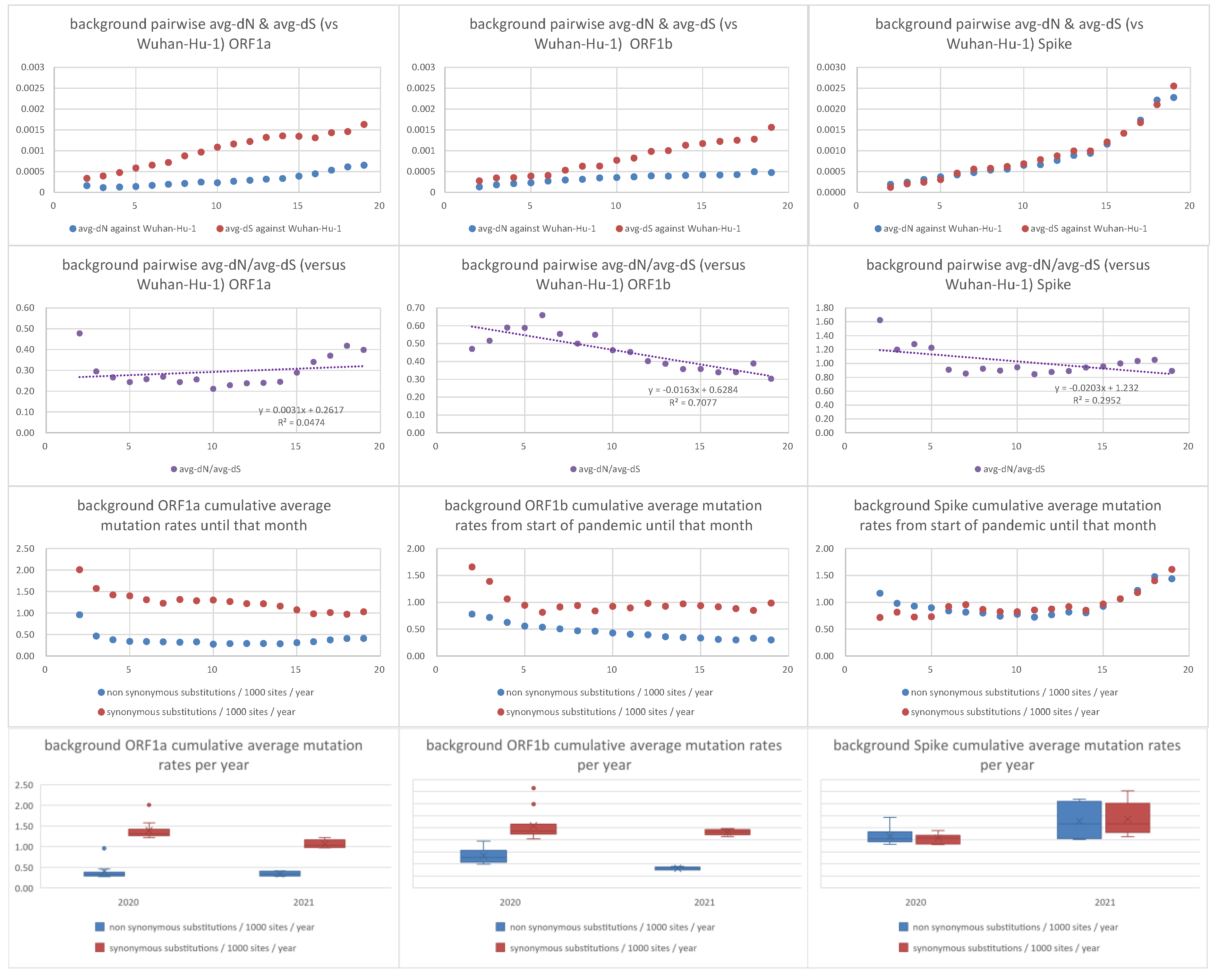
Figure 4. Pairwise average dN, dS, dN/dS, synonymous and non-synonymous mutation
rates of background non-VoC/VoI lineages against Wuhan-Hu-1 strain. The x-axis in
the first nine graphs denotes number of months from the beginning of the pandemic
(December 2019).
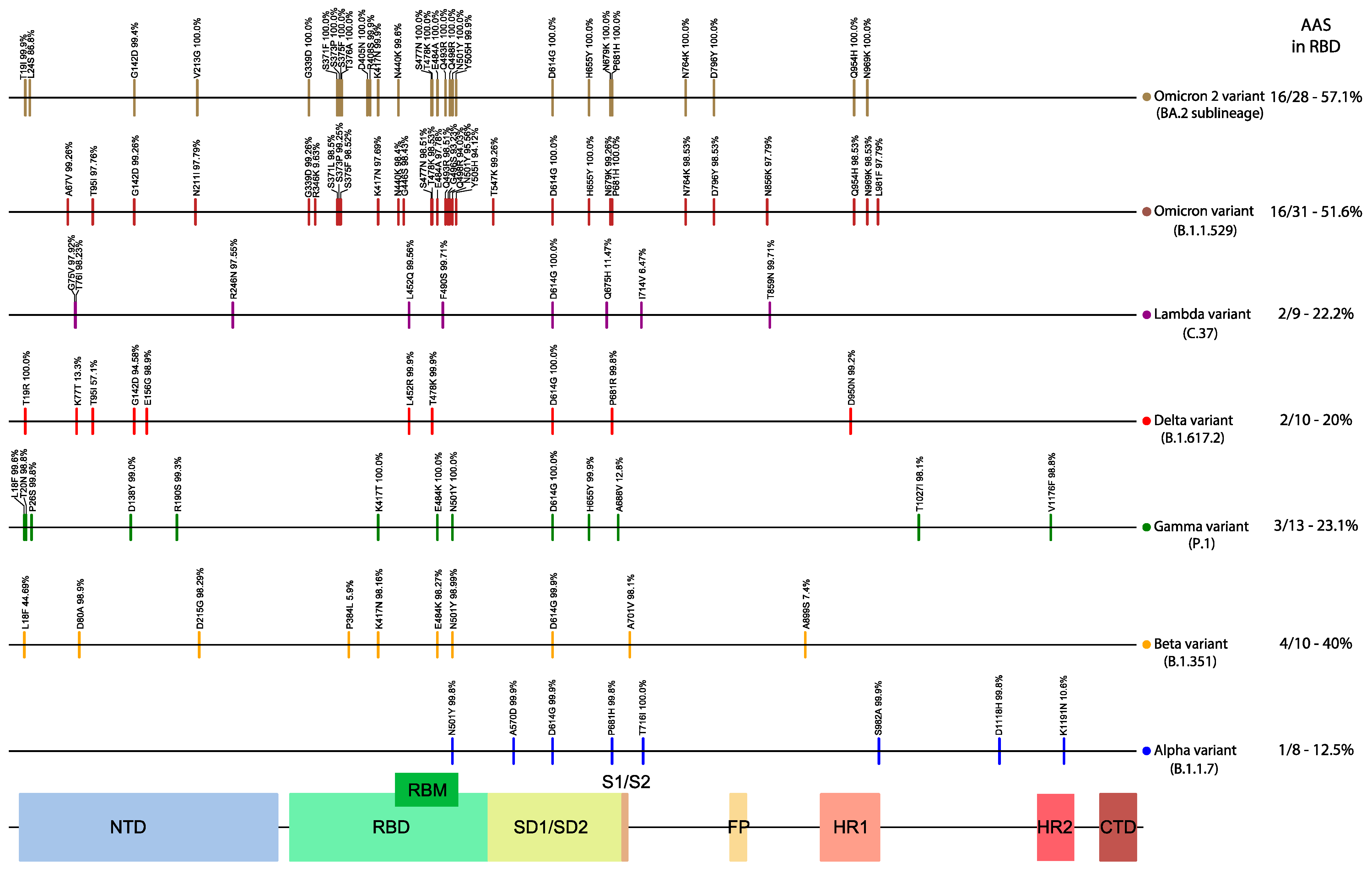
Figure 5. Amino acid substitutions (AAS) of the selected variant lineages (compared
to Wuhan-Hu-1), across the Spike. The observed frequency of each AAS for that
lineage is also displayed above the corresponding vertical bar. On the right side is
the number of AAS in RBD and Table 1 sequence. NTD: N-terminal domain; RBD:
receptor-binding domain; RBM: receptor-binding motif.
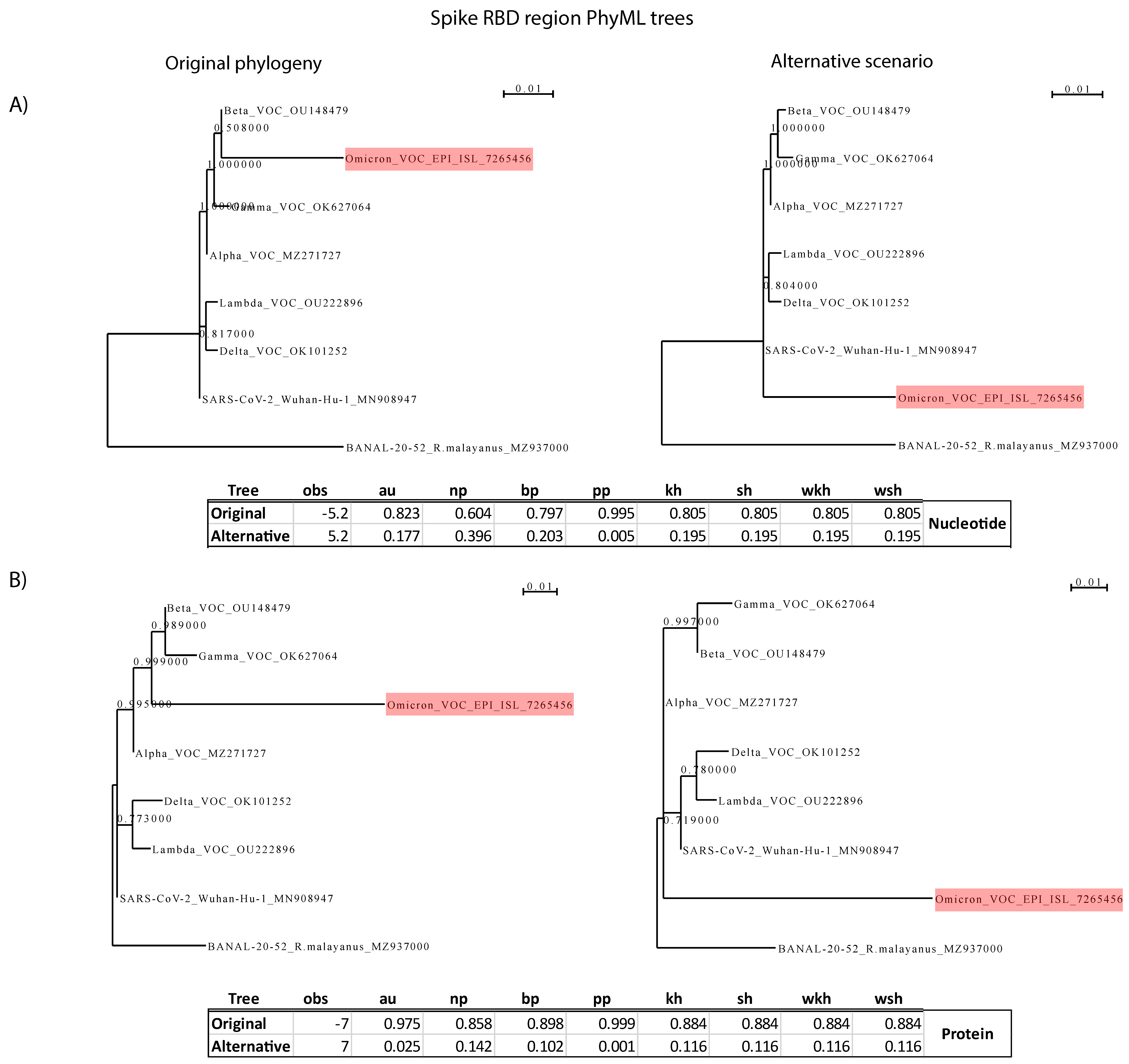
Figure 6. CONSEL analysis for the Spike RBD. (A) Analysis based on RBD
nucleotide sequences. (B) Analysis based on RBD protein sequences. On the
left side is the null hypothesis of RBD divergence by accumulation of point
mutations of an existing SARS-CoV-2 lineage; on the right is Scheme 2. The branch
lengths of the alternative hypothesis tree were optimized by PhyML. No analysis
favors the alternative hypothesis of recombination with a closely related
Sarbecovirus.
The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination:
Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2
Coronaviruses (CoVs) constitute a large and diverse subfamily of positive-sense
single-stranded RNA viruses. They are found in many mammals and birds and have great
importance for the health of humans and farm animals. The current SARS-CoV-2 pandemic, as
well as many previous epidemics in humans that were of zoonotic origin, highlights the
importance of studying the evolution of the entire CoV subfamily in order to understand how
novel strains emerge and which molecular processes affect their adaptation,
transmissibility, host/tissue tropism, and patho non-homologous genicity. In this review, we
focus on studies over the last two years that reveal the impact of point mutations,
insertions/deletions, and intratypic/intertypic homologous and non-homologous recombination
events on the evolution of CoVs. We discuss whether the next generations of CoV vaccines
should be directed against other CoV proteins in addition to or instead of spike. Based on
the observed patterns of molecular evolution for the entire subfamily, we discuss five
scenarios for the future evolutionary path of SARS-CoV-2 and the COVID-19 pandemic. Finally,
within this evolutionary context, we discuss the recently emerged Omicron (B.1.1.529) VoC.
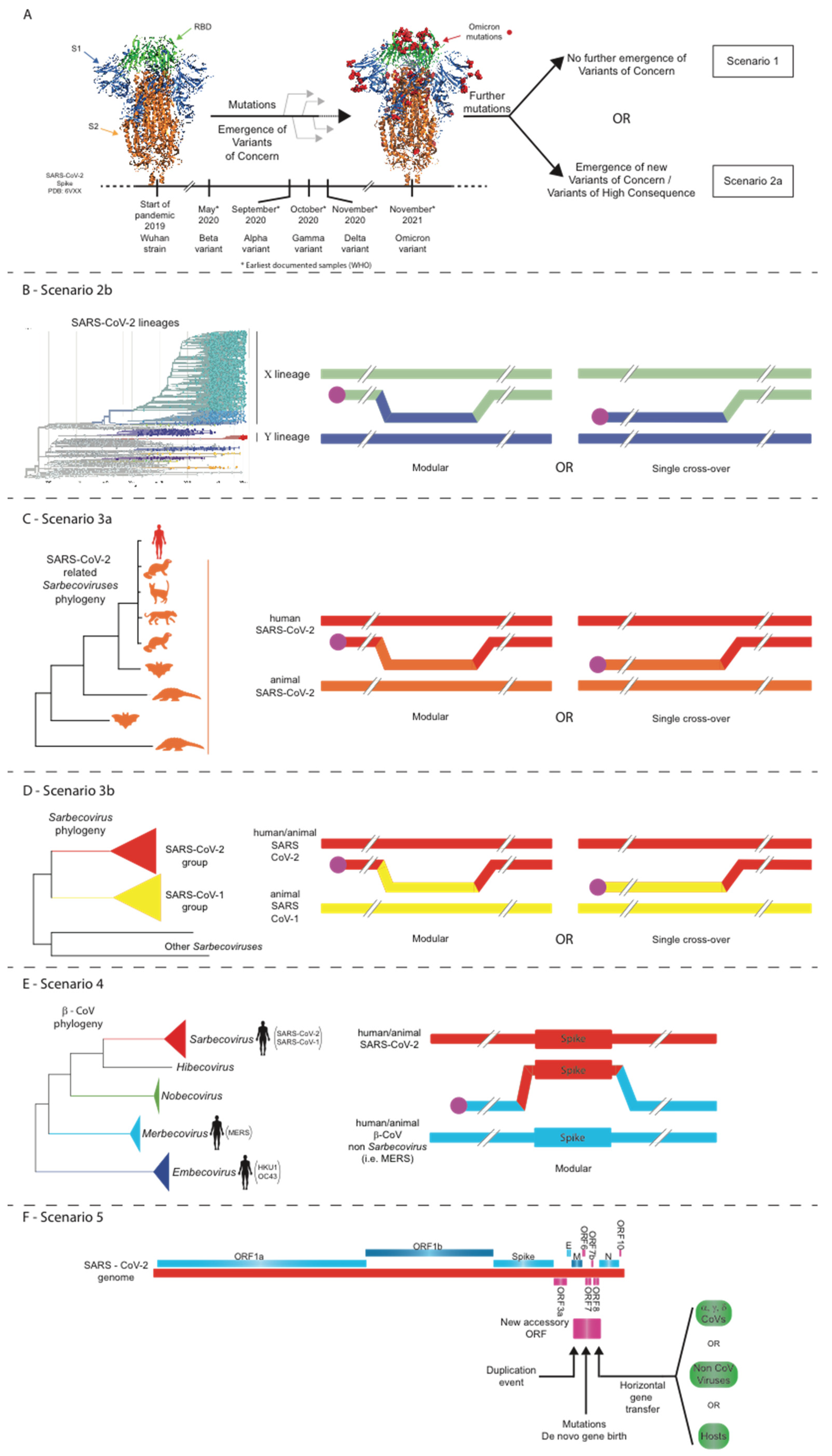
Figure 1. Five scenarios for the future evolutionary trajectory of SARS-CoV-2.
(A) Scenario 1: structural constraints limit any further evolution of the
SARS-CoV-2 spike; Scenario 2a: point mutations, insertions/deletions, and/or
intra-SARS-CoV-2 recombination events lead to the evolution of novel SARS-CoV-2
strains. (B) Scenario 2b: intra-SARS-CoV-2 recombination events lead to the
evolution of novel SARS-CoV-2 strains. (C) Scenario 3a: intratypic
recombinations between SARS-CoV-2 and closely related sarbecoviruses. (D)
Scenario 3b: intratypic recombinations between SARS-CoV-2 and other related
sarbecoviruses. (E) Scenario 4: intertypic recombination between SARS-CoV-2
and viruses from other Beta-CoV subgenera. (F) Scenario 5: non-homologous
recombination of SARS-CoV-2 with other coronaviruses or even other viruses/hosts.